NEWS
LATEST UPDATES
-

30.03.2026
Identifican el papel del gen BAP1 en el desarrollo anómalo de la placenta asociado a la preeclampsia precoz.
-
27.02.2026
El Conseller de Sanidad, Marciano Gómez, clausura en el CIPF la X Jornada AITER
-
19.02.2026
Conferencia de Mª Jesús Vicent en la Pobla Llarga
-
09.02.2026
Un grupo de investigación del CIPF descubre una proteína clave en la reparación del cerebro tras un traumatismo
-
30.01.2026
El CIPF se suma a la iniciativa STEM Talent Girl para despertar vocaciones científicas
-
06.02.2026
El CIPF y VRAIN celebran su primera jornada para promover proyectos de investigación conjuntos
-
05.12.2025
Nanoestrellas de oro que activan fármacos con luz: un nuevo enfoque contra el cáncer
-

25.11.2025
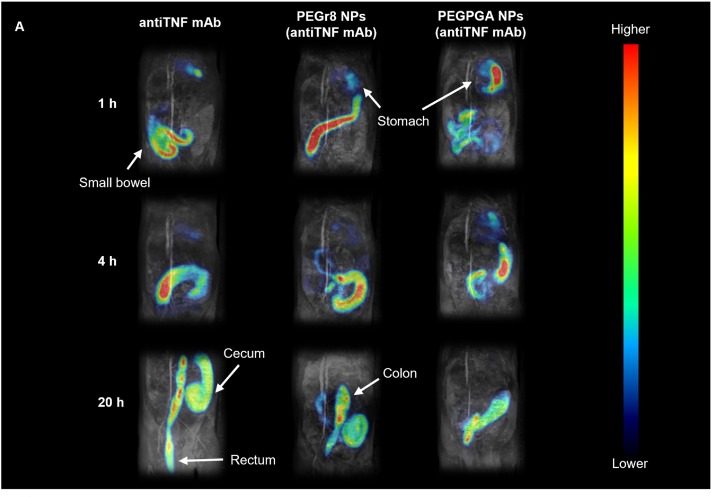
El CIPF participa en el desarrollo de una nueva plataforma nanotecnológica para la administración oral de fármacos biológicos
-
31.10.2025
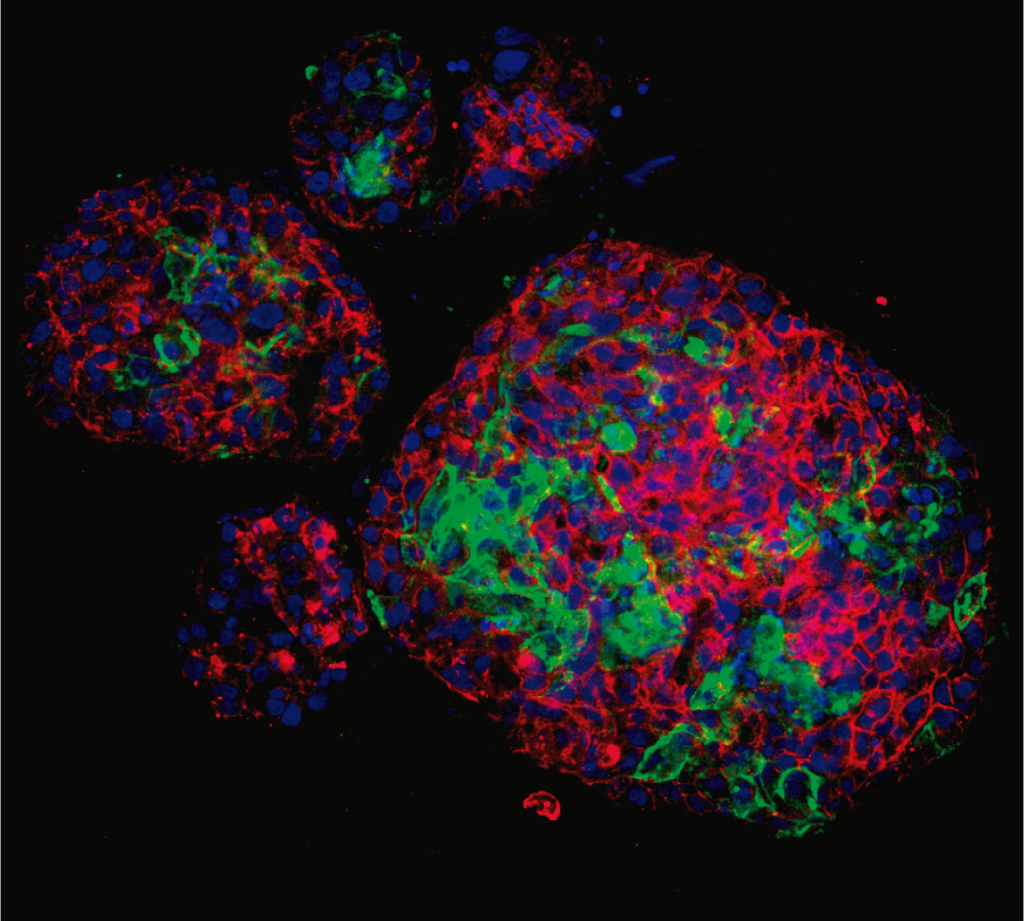
El CIPF lidera un estudio innovador con potencial terapéutico en oncología y enfermedades autoinmunes
-

23.10.2025
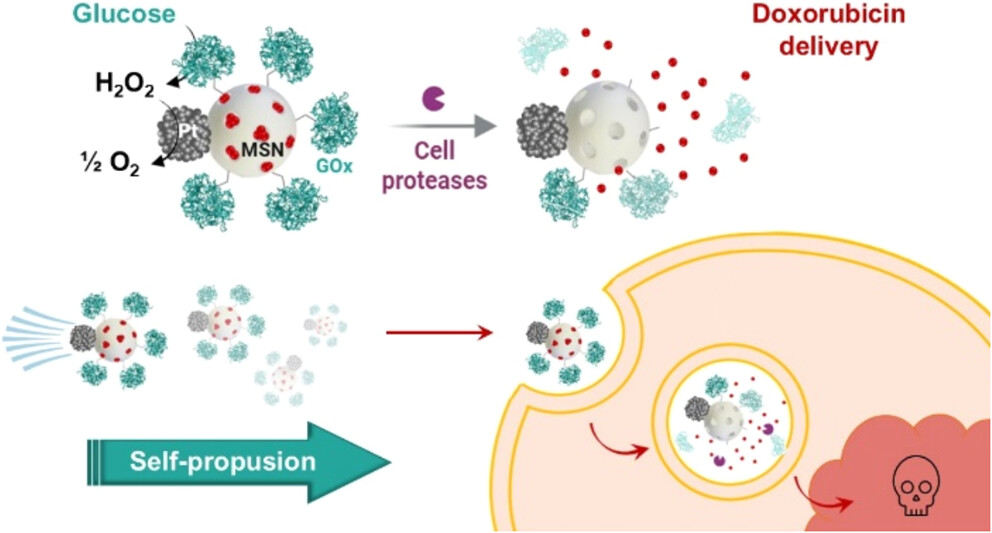
Nanomotores que usan glucosa como “gasolina” para combatir el cáncer